
将生成式预训练与推理时计算扩展统一于全原子框架
[size=1em]核心信息 - • 来源:Didi, K., Zhang, Z., Zhou, G., Reidenbach, D., Cao, Z., Cha, S., ... & Kreis, K. Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute. ICLR 2026
- • 机构:NVIDIA、牛津大学、Mila、首尔国立大学等
一、研究背景与核心问题1.1 蛋白质结合体设计的重要性蛋白质-蛋白质相互作用(PPI)是几乎所有生命活动的分子基础。设计能以高亲和力与特定靶标结合的从头蛋白质(de novo binder),是现代计算生物学中最重要的挑战之一,其应用涵盖: - • 治疗性抗体与蛋白质药物(靶向 TNF-α、PD-L1、VEGFA 等)
- • 疫苗设计(靶向病毒蛋白表面抗原)
- • 诊断试剂与传感器
- • 酶工程与代谢通路改造
随着 AlphaFold2 等结构预测工具的成熟,基于结构的从头设计(structure-based de novo design)已成为主流方向。 1.2 现有方法的两大流派及其局限当前 AI 驱动的结合体设计方法形成了两个相互割裂的技术路线: 生成式方法(Generative Methods)代表:RFDiffusion、Protpardelle-1c、APM 这类方法将结合体设计建模为条件生成问题:在大量结合复合物(binder-target complex)结构上训练扩散或流匹配模型,推理阶段直接从靶标结构出发生成结合体。 优势:推理速度快,具有从训练数据中习得的结构先验,一次生成即可产生合理的候选结构。 劣势: - • 生成质量受限于训练数据规模与多样性
- • 无法在推理阶段动态地利用外部评分信号优化候选
- • 早期方法(RFDiffusion)只生成主链骨架,需要额外的序列重设计步骤(ProteinMPNN / LigandMPNN)
幻觉式方法(Hallucination Methods)代表:BindCraft、BoltzDesign、AlphaDesign 这类方法不训练生成模型,而是将结构预测模型(AlphaFold2、Boltz-1)的置信度分数和对齐误差作为可微分的损失函数,通过反向传播直接优化氨基酸序列。 优势:优化目标与最终评估标准高度一致,无需额外的序列重设计步骤。 劣势: - • 计算代价极高(每个候选都需要反复调用 AlphaFold2)
- • 序列空间的梯度优化需要复杂的离散化近似(softmax 退火、straight-through estimator)
- • 没有生成先验约束,优化轨迹容易发散或陷入局部最优
- • 缺乏全局搜索能力,本质上是局部优化
1.3 问题的核心矛盾作者指出,上述二元对立与大语言模型(LLM)、图像生成等领域的发展路径形成了鲜明对比: [size=1em]现代 AI 系统同时扩展训练数据(training-time compute)和推理计算(inference-time compute)。当前蛋白质设计方法,要么只做前者,要么只做后者。
具体类比如下: AI 系统预训练阶段推理优化阶段
现代 LLM(如 o1、GPT-4)大规模语料预训练链式思维、MCTS、Best-of-N
图像生成(Stable Diffusion)海量图文对训练分类器引导、奖励函数引导
生成式蛋白质设计复合物结构训练❌ 无推理时优化
幻觉式蛋白质设计❌ 无生成先验AlphaFold2 梯度等优化
Proteína-Complexa(本文)✅ 大规模预训练✅ 多种推理时扩展算法这一洞察正是本文的核心动机。
二、Proteína-Complexa 框架总览Complexa 由三个相互配合的核心模块构成: - 1. Teddymer:大规模合成结合体-靶标数据集(解决数据瓶颈)
- 2. 基础生成模型:扩展 La-Proteína 至条件生成场景(提供强生成先验)
- 3. 推理时计算扩展:多种搜索算法在生成过程中引入外部奖励(统一两大范式)
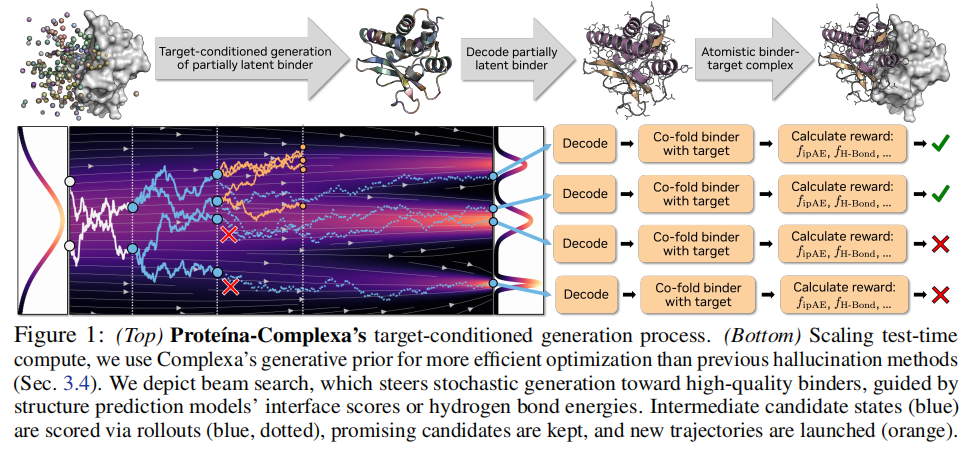
三、Teddymer:从单体域相互作用挖掘大规模训练数据3.1 数据瓶颈问题训练一个高质量的结合体生成模型,需要大量成对的结合体-靶标复合物结构。然而: - • 蛋白质数据库(PDB) 中实验解析的多链复合物约 22.5 万条,经严格质量过滤后仅余约 4.5 万条可用二聚体
- • AlphaFold Database(AFDB) 提供了超 2 亿条计算预测的单体结构,但没有相应的复合物数据库
现有生成式方法(如 RFDiffusion)受限于此,在多样性和泛化能力上都有明显天花板。 3.2 核心洞察:域间相互作用 ≈ 链间相互作用作者基于一个关键的生物物理假设: [size=1em]多域单体蛋白内,相邻结构域之间的物理接触界面,在本质上与多链蛋白复合物中链间的结合界面具有相似的生物物理特性。
这一假设在文献中有先例(Sen & Madhusudhan, 2022),并在本文中通过界面统计分析得到验证:Teddymer 与 PDB 多链复合物在界面氢键数、疏水性、形状互补性、埋藏溶剂可及表面积(dSASA)等多项指标上的分布高度重叠。 3.3 构建流程AFDB(2亿条单体结构)
↓ 筛选 AFDB50(50% 序列同一性聚类代表)
47,180,623 条结构
↓ 应用 TED 域注释,拆分为多域 → 多链格式
↓ 提取所有链对,以 Cα-Cα 距离 ≤ 10Å 且至少 4 个残基为空间邻近标准
123,606,001 条候选二聚体
↓ 过滤:双链均具有 CATH C.A.T. 级别注释
10,089,503 条二聚体
↓ 使用 GPU 加速的 Foldseek-Multimer 进行聚类
↓ 链级结构相似度阈值 0.7,界面 lDDT 阈值 0.3
3,556,223 个聚类
↓ 进一步过滤:界面长度 > 10,界面 pAE < 10,界面 pLDDT > 70
510,454 个代表结构(最终训练集)3.4 数据规模对比数据集来源可用训练样本
Filtered PDB Multimers实验解析~45,856
PLINDER(蛋白质-配体)实验解析~78,437
Teddymer(本文)合成构建~510,454
AFDB 单体(用于预训练)计算预测~588,318Teddymer 将可用的成对蛋白质-蛋白质结合数据扩展了约 11 倍,且成本仅为数据处理的计算消耗。
四、基础生成模型:全原子条件生成4.1 技术基础:La-Proteína 框架回顾Complexa 建立在 La-Proteína(Geffner et al., 2026,同组前作)之上,该框架的核心思想是部分隐空间流匹配(Partially Latent Flow Matching): 表示层面:将蛋白质分解为两个部分: - • 显式部分:α 碳坐标 ,在三维空间中直接建模
- • 潜在部分:通过 VAE 编码器将序列 和非 α 碳原子坐标 编码为固定维度连续潜在变量
生成层面:对 联合进行流匹配(Rectified Flow 公式化):其中 分别是 α 碳坐标和潜在变量的独立插值时间,采用不同的噪声调度(α 碳使用指数调度,潜变量使用二次调度),这对高质量全原子生成至关重要。架构层面:使用纯 Transformer 架构(Pair-Biased Attention),避免了 AlphaFold 中的三角更新层(Triangular Multiplicative Update),大幅提高了计算效率和可扩展性。 4.2 靶标条件化:隐空间靶标条件机制将 La-Proteína 扩展至结合体设计,需要模型在生成结合体时感知靶标结构。本文提出的隐空间靶标条件化(Latent Target Conditioning)机制如下: 靶标表征:使用 Atom37 表示(每个残基最多 37 个原子的三维坐标)+ 氨基酸身份独热编码 + 二值热点 token(标记靶标上应靠近结合体的界面残基)。 条件注入:靶标特征经线性嵌入后,在 token 维度与结合体的 表示直接拼接,形成联合序列表示:带噪结合体干净靶标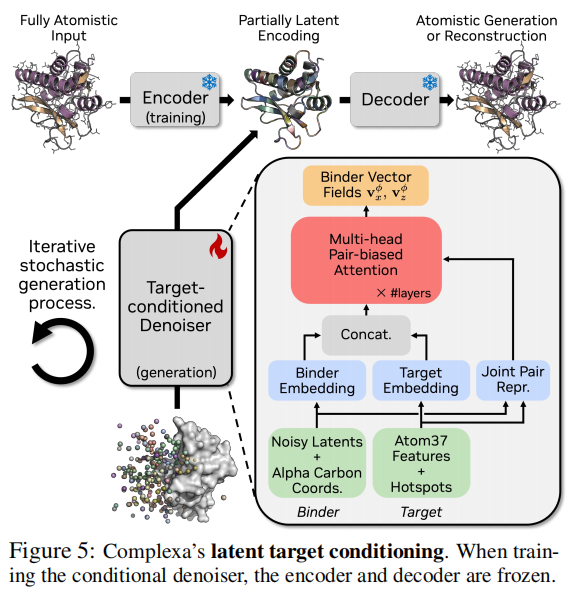 关键设计:VAE 的编解码器仅处理单体蛋白,条件化仅施加在流匹配去噪器上。这意味着无论靶标是蛋白质还是小分子,VAE 组件无需修改,同一套自编码器在所有下游任务中复用,极大简化了系统复杂度。 Pair 表示:去噪器使用静态 pair 表示捕获残基对间的结构关系,包含: - • 结合体内部 pair:Cα 距离(1Å bin,1-30Å)+ 序列分离距离
- • 靶标内部 pair:骨架原子距离 + 链索引(多链靶标)+ 热点 pair 变量
- • 结合体-靶标跨链 pair:噪声 Cα 与靶标骨架原子之间的距离
小分子靶标的扩展:对于小分子(配体),在原子级别直接表征:原子类型独热编码 + 三维坐标 + 原子电荷 + 图拉普拉斯位置编码(graph Laplacian positional encoding)+ 原子名称独热编码,并将分子内部键序和键掩码引入 pair 表示。这使得同一框架可以无缝处理蛋白质和小分子两类靶标。 4.3 平移噪声:解决全局定位的隐性捷径问题这是本文一个精巧但关键的技术细节。 问题发现:在标准流匹配中,对结合体 Cα 坐标进行插值 ,若训练时将靶标置于原点,则中间状态的质心为 。模型可以利用此关系,在 时直接从 恢复结合体的真实质心,完全绕过了学习"如何将结合体定位至靶标界面"这一核心能力。解决方案:引入全局平移噪声 ( nm),修改插值为:Fourier 视角:从频域角度理解,扩散/流匹配模型在生成过程早期主要处理低频分量。全局平移对应最低频模式,加入平移噪声相当于强制模型在整个去噪过程中持续精化结合体的全局位置,而不只是在早期决定。 消融实验证明,去掉平移噪声后,在 19 个靶标上的平均成功率几乎归零。 4.4 分阶段训练策略借鉴 LLM 的预训练-微调范式,采用多阶段训练: 阶段 1:VAE 预训练
数据:AFDB 单体(∼50 万条,pLDDT > 80,长度 32-256)
设备:16×A100,500k 步
目标:学习通用的蛋白质全原子编解码能力
阶段 2:VAE 微调(引入 PDB 真实结构)
数据:PDB 单链(∼11 万条,长度 50-256,分辨率 < 5Å)
原因:AFDB 结构过于理想化(由折叠模型生成),加入真实晶体学数据
提升解码器对真实构象多样性的覆盖
阶段 3:流匹配模型预训练(单体生成)
数据:AFDB Foldseek 聚类代表(∼58 万条)
设备:32×A100,540k 步
目标:习得通用蛋白质结构生成的流场
阶段 4:流匹配模型微调(结合体条件生成)
数据:Teddymer + PDB 多链(8:2 混合比例)
设备:96×A100,290k 步
目标:学习给定靶标条件下生成结合体的能力
(小分子靶标变体):
阶段 3b:LoRA 微调
数据:PLINDER + AFDB 单体(50% 目标 dropout 防止过拟合)
设备:96×A100,60k 步模型规模:去噪器约 1.59 亿参数,VAE 约 2.56 亿参数,序列表示维度 768,pair 表示维度 256,14 层 Transformer,12 个注意力头。
五、推理时计算扩展:四大算法统一两大范式5.1 成功度量标准在介绍算法之前,需要了解评判成功的标准: 蛋白质靶标(遵循 AlphaProteo 标准,使用 ColabDesign-AF2 重折叠验证): - • (折叠置信度)
- • Å(界面预测对齐误差)
- • Å(结合体自洽 RMSD)
小分子靶标(使用 RosettaFold-3 重折叠验证): 所有成功设计进一步用 FoldSeek 聚类,报告独特成功数(unique successes),避免重复计算高度相似的结构。 5.2 Best-of-N 采样最简单的推理时扩展:独立生成 个样本,选取满足成功标准的所有样本。实践中 最多扩展至 51,200,生成以批模式运行,折叠评估以单样本模式运行。结果表明,对于容易靶标,此方法已显著超越所有幻觉基线方法。5.3 束搜索(Beam Search)维护 条并行去噪轨迹(束宽 ),每次推进 步后 branching 出 条候选(分支因子 ):关键实现细节: - • 不使用 Tweedie 公式从噪声中间态估计奖励(因结构预测模型对噪声序列不可靠),而是将所有候选状态直接 rollout 至完全去噪的干净样本,用折叠模型评分
- • rollout 过程中产生的所有满足成功标准的样本均加入结果集,而不只保留最终选定的束
- • 整个搜索过程可以重复多轮,直至达到算力预算上限
每步搜索涉及 次完整 rollout,加上折叠模型评估,计算量有限,因为 Complexa 的生成器本身足够快速。5.4 Feynman-Kac Steering(FKS)基于 Feynman-Kac 相互作用粒子系统(FK-IPS)的引导方法,目标是从以下倾斜分布中采样: 与束搜索的关键区别在于:束搜索使用硬性 top-N 选择,而 FKS 使用重要性采样: 这种软性选择保留了更多样本多样性,避免过早收敛到单一模式。在实践中,FKS 与束搜索在大多数靶标上性能相当,各有擅长场景。 5.5 Monte Carlo Tree Search(MCTS)将整个去噪过程建模为搜索树,每个节点对应一个中间噪声状态 ,搜索树中的不同路径对应不同的随机去噪轨迹。节点选择标准(改进 UCB 公式): 开发探索其中 为节点访问计数, 为探索常数。连续状态空间的适配:标准 MCTS 假设离散有限动作空间,但流匹配的状态空间是连续无界的。本文的解决方案: - • 以概率 随机扩展新子节点(运行 步去噪生成新子状态)
- • 以概率 选择已有最优子节点继续探索
- • 这相当于一个"软性分支因子":若节点被访问 次,期望有 个子节点
批推理兼容性:由于同一个 参数应用于批内所有节点,确保同一批次内的所有去噪步骤处于相同时间戳,从而支持 GPU 批量推理,不牺牲计算效率。实践超参:,,,(每次决策步执行的模拟次数)。5.6 Generate & Hallucinate(G&H):两大范式的直接混合最简洁的组合方案: - 1. 用 Complexa 生成候选结合体序列(利用生成先验得到合理初始点)
- 2. 用 BindCraft 的第 4 阶段(离散突变优化)进行局部序列精炼
为何跳过 BindCraft 的前三个阶段(梯度 logit 优化)? 作者给出了深刻的分析:对于容易靶标,Complexa 生成的序列已经是高质量候选,logit 优化反而浪费计算资源且引入不必要的扰动。早期的梯度优化阶段本质上是"将随机初始序列快速推向合理区域",但 Complexa 已经完成了这一步。因此,直接从生成序列出发进行精准的离散突变,性价比更高。对于困难靶标,才考虑加入部分 logit 优化阶段。 5.7 界面氢键优化这是 Complexa 框架独特的能力,体现了其奖励函数的灵活性。 蛋白质-靶标之间的强相互作用通常需要大量界面氢键。本文将氢键能量(通过 Rosetta 能量函数计算,使用 GPU 加速的 tmol 实现)引入推理时优化: 结果(在 19 个靶标平均): 奖励配置独特成功数平均界面氢键数
无奖励(Best-of-N 等价)77.005.27
仅 ipAE83.365.52
仅 H-Bond82.367.15
ipAE + H-Bond86.266.52这表明物理能量函数与预测模型置信度可以协同优化,且优化氢键并不以牺牲折叠置信度为代价。事实上,两者存在正相关:降低 ipAE 分数与增加氢键数之间的 Spearman 相关系数为 。
六、实验评估:全面的基准测试6.1 评测协议- • 蛋白质靶标:22 个靶标(来自 AlphaProteo 10 个 + BindCraft 12 个),每个靶标生成 200 个候选结构(长度 40-250 残基)
- • 小分子靶标:4 个小分子(SAM、OQO、FAD、IAI),每个生成 200 个候选(长度 100 残基)
- • 序列评估:对全原子模型,分别评估自生成序列(Self)、ProteinMPNN 重设计序列(MPNN)、固定界面残基重设计序列(MPNN-FI)
- • 折叠验证:蛋白质靶标使用 ColabDesign-AF2,小分子靶标使用 RosettaFold-3
6.2 生成式基线比较(无推理时优化)蛋白质靶标(表 2): 方法独特成功数(Self)独特成功数(MPNN-FI)独特成功数(MPNN)生成时间(s)新颖性
RFDiffusion——4.6870.80.87
Protpardelle-1c——0.738.130.77
APM0.311.523.1573.10.86
Complexa(本文)9.1013.614.415.60.80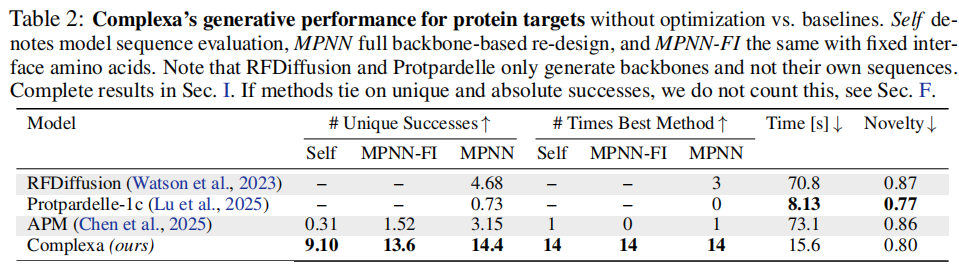 • Complexa 自生成序列(9.10)超过所有竞争对手的 MPNN 重设计结果(最好 4.68) • Complexa 自生成序列(9.10)超过所有竞争对手的 MPNN 重设计结果(最好 4.68)- • 采样速度比 RFDiffusion 和 APM 快约 4-5 倍
- • 新颖性(TM-Score 对 PDB 数据库)0.80,具有良好的序列和结构新颖性
小分子靶标(表 1): 方法SAMOQOFADIAI时间(s)
RFDiffusion-AllAtom235887.4
Complexa(本文)106171913.5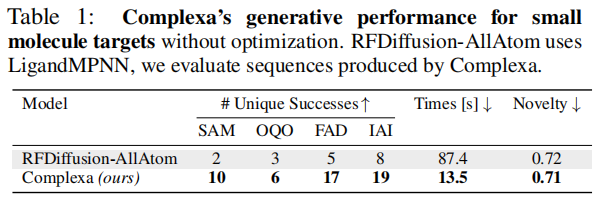 Complexa 在所有四个小分子靶标上均以大幅度超越唯一的公开竞争对手,同时采样速度提升约 6.5 倍。 6.3 推理时扩展 vs 幻觉方法(图 7-9)在归一化的 GPU 小时预算下,与 BindCraft、BoltzDesign、AlphaDesign 进行比较: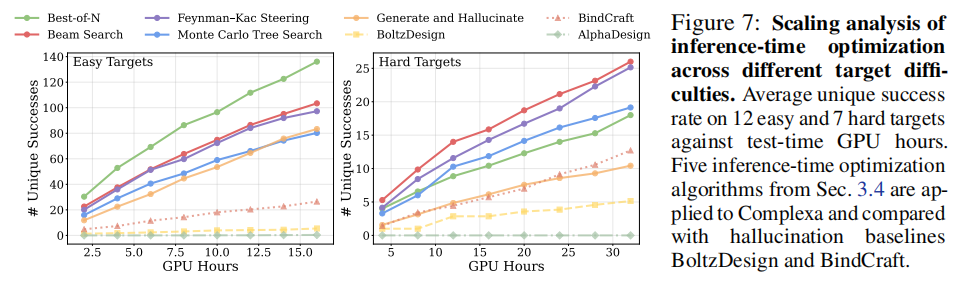 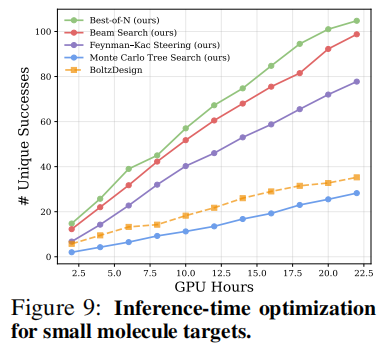 容易靶标(12 个): 容易靶标(12 个):- • 简单的 Best-of-N 已在所有计算预算下超越三个幻觉基线
- • Beam Search 在 15 GPU 小时时达到约 130 个独特成功,相比之下 BindCraft 和 BoltzDesign 约 20 个
困难靶标(7 个): - • Best-of-N 提升有限(约 5 个),需要 Beam Search、FKS、MCTS 的结构化搜索
- • 在 30 GPU 小时下,Complexa 最佳方法约 22 个独特成功,BindCraft/BoltzDesign 约 5-8 个
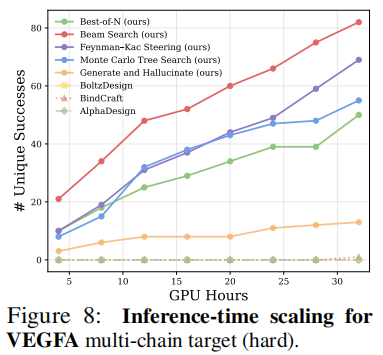
极困难多链靶标(TNF-α、H1、IL17A): 这三个靶标对所有方法都极具挑战性,AlphaFold2-Multimer 置信度往往较低。针对这些靶标,优化策略进行了调整:奖励函数改为 归一化ipAE + pLDDT 的组合,MCTS 后接 G&H 的局部精炼(每轮突变率提高至 5% 结合体长度)。 结果:在超过 100 GPU 小时的搜索后: - • TNF-α(三链靶标):15 个独特成功(475 GPU 小时)
- • H1(两链靶标):7 个独特成功(604 GPU 小时)
- • IL17A(两链靶标):1 个独特成功(387 GPU 小时)
所有幻觉基线在 32 GPU 小时内均无任何成功。 6.4 酶设计基准在 AME(Atomic Motif Enzyme)基准的 41 个设计任务上: 方法成功任务数(自生成序列)成功任务数(LigandMPNN × 8)
RFDiffusion230 / 4130 / 41
Complexa(本文)41 / 4140 / 41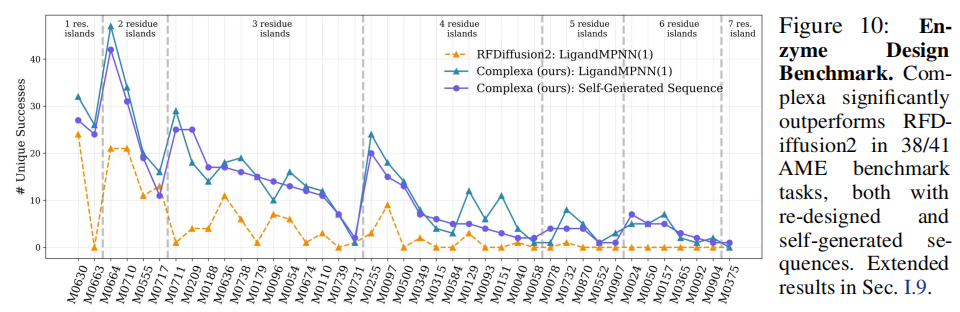 Complexa 在 38/41 个任务上超越 RFDiffusion2(best-of-8 LigandMPNN 条件),且在所有含 ≥4 个残基岛(residue islands)的任务上均胜出。这展示了框架在复杂的催化活性位点重建任务中的泛化能力。 6.5 折叠多样性控制:CAT 标签条件化利用 CATH 分类(CAT 标签)对结合体的二级结构偏好进行显式控制: - • Mainly Alpha:主要生成 α 螺旋结合体
- • Mainly Beta:主要生成 β 折叠结合体
- • Mixed Alpha Beta:混合结构
这解决了现有蛋白质生成模型普遍存在的"α 螺旋偏差"问题,显著提高了成功结合体的结构多样性。在多个靶标(BHRF1、Derf21、IFNAR2、PD-1、PD-L1)的可视化结果显示,不同 CAT 条件下生成的结合体在拓扑结构上显著不同,且均满足成功标准。 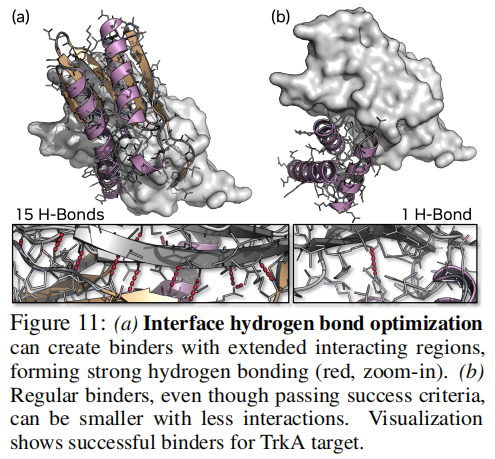 七、消融研究7.1 Teddymer 数据的必要性 七、消融研究7.1 Teddymer 数据的必要性在去掉 Teddymer(仅使用 PDB 多链数据训练)后,在 19 个靶标上的性能变化: 评估方式Complexa(完整)Complexa(无 Teddymer)下降幅度
MPNN14.43.84-73%
MPNN-FI13.51.68-88%
Self9.100.15-98%去掉 Teddymer 后,自生成序列的成功率几乎归零(19 个靶标中有 0 个是最佳方法),证明 Teddymer 提供的大规模多样化蛋白质-蛋白质相互作用样本是模型泛化能力的根本来源,仅靠过滤后的 PDB 数据(约 4.5 万条)远远不足。 此外,论文还用 RosettaFold-3 和 Boltz-2 进行了独立验证(避免与 AlphaFold2 系同源偏差的质疑),结论一致:有 Teddymer 的模型在第三方折叠模型评估下依然显著领先。 7.2 平移噪声的必要性评估方式Complexa(完整)Complexa(无平移噪声)
MPNN14.43.73
MPNN-FI13.53.89
Self9.101.47去掉平移噪声后,性能同样大幅下降,验证了该设计对模型学习全局定位能力的关键作用。少数困难靶标在去掉平移噪声后略有提升,作者认为这是因为额外的噪声使本已困难的任务更难学习,但整体上平移噪声是有益的。
八、技术创新总结与深层分析8.1 方法论层面的根本突破本文最重要的贡献不是某个具体的技术细节,而是确立了一个新的方法论范式:将蛋白质设计问题的解决框架与现代大规模生成模型的最佳实践对齐。 这种对齐体现在三个层次: - 1. 数据扩展:通过 Teddymer 突破实验数据瓶颈,类比 LLM 的网络爬虫数据
- 2. 模型预训练:分阶段训练策略,类比 LLM 的预训练-RLHF 流程
- 3. 推理时优化:将 Best-of-N、Beam Search、MCTS 等推理策略系统引入蛋白质设计
8.2 框架设计的工程优雅性- • 同一 VAE 用于所有靶标类型:架构解耦使得扩展到新靶标模态时只需微调流匹配模型,不需重新训练整个系统
- • 纯 Transformer 架构:避免了 AF2 的三角更新层,保持线性而非二次的内存增长,使得大批量推理(用于 Best-of-N)在实际中可行
- • 独立的 、 时间调度:允许 α 碳坐标(低频、全局结构)在潜变量(高频、侧链细节)之前先收敛,符合蛋白质折叠的层次性特征
8.3 推理时算法的适配挑战将扩散/流匹配文献中的推理时优化算法(Fernandes et al., Singhal et al., Yoon et al.)迁移到蛋白质设计并非平凡工作,主要挑战有: - 1. 噪声中间态不可直接评分:蛋白质结构预测模型只对真实序列可靠,对噪声中间态完全失效,因此必须 rollout 到干净样本再打分(不能用 Tweedie 估计)
- 2. 连续无界状态空间:MCTS 的传统离散分支因子在连续流场中失效,需要随机扩展策略
- 3. 批推理同步约束:MCTS 中不同路径可能处于不同时间步,破坏 GPU 批并行。本文通过统一 参数确保批内时间步同步
- 4. 计算效率:每次奖励评估都涉及完整的折叠模型调用(ColabDesign-AF2),是主要瓶颈。Complexa 的快速生成器使得每步搜索的 rollout 代价相对可控
九、局限性与未来方向9.1 论文明确指出的局限- • 缺乏湿实验验证:所有结果均为计算机内(in-silico)评估,实际结合亲和力和特异性需要实验验证
- • 单一靶标模态:目前蛋白质靶标和小分子靶标分别用独立模型处理,尚未统一
- • 未覆盖的分子模态:DNA、RNA、多肽、抗体等尚未纳入框架
9.2 深层的技术挑战- • 奖励黑客(Reward Hacking)问题:推理时优化针对 ipAE 分数,是否会产生"欺骗"折叠模型但实际不结合的设计?作者初步分析显示 ipAE 与氢键数负相关,未见明显对抗优化,但更严格的实验验证仍然必要
- • 长程搜索的饱和:在 SpCas9 案例研究中,800+ GPU 小时后独特成功数增长开始放缓,表明生成模型能覆盖的解空间有限
- • 小分子结合位点保留:当前的奖励函数(min ipAE)不能完全捕获配体结合位点守恒,导致 ligand scRMSD 成为主要失败模式
9.3 最令人期待的未来方向统一多模态生成模型:训练单一模型同时处理蛋白质、肽段、小分子、核酸、抗体等多种分子模态作为靶标或生成对象,跨模态迁移学习有望进一步提升所有任务的性能(参考 UniMoMo, Kong et al., 2025)。 引入生物物理约束:将热稳定性、特异性、可溶性等分子性质的预测模型集成为额外奖励信号,实现多目标推理时优化。 主动学习闭环:将湿实验反馈(实测结合亲和力)循环整合为新的训练信号,实现生成模型与实验的迭代共进化。
[size=1em]结语 [size=1em]Proteína-Complexa 代表的不只是一个性能更好的设计工具,而是蛋白质设计方法论与现代生成 AI 范式的一次深度对齐。这种对齐——大规模数据预训练 + 推理时计算扩展——在 NLP 和图像生成领域已经被反复验证为提升系统能力的普适路径。将其引入计算蛋白质设计,可能正在开启这一领域新的发展曲线。 [size=1em]对于关注 AI for Science、计算生物学、或生成模型在科学中应用的研究者,这篇文章值得深入阅读和持续关注。
|
 关注公众号
关注公众号
 发表于 2026-3-22 22:18:58
发表于 2026-3-22 22:18:58
