|
无序蛋白在细胞中发挥着广泛而重要的功能,但由于其高度动态和结构异质性,对其结构进行精确表征一直是结构生物学中的重大挑战。本文提出了一项由研究社区推动的倡议,旨在建立一个用于确定无序蛋白构象系综的统一框架。该框架通过整合先进的实验技术与计算方法,包括基于知识的采样、增强分子动力学模拟以及机器学习模型,从而实现对无序蛋白多构象状态的系统刻画。整个框架由三个相互关联的模块组成:实验数据获取、构象系综生成以及结果验证与比较。研究人员认为,通过标准化流程并推动社区协作,可以实现对无序蛋白构象系综更加准确和可重复的解析,并促进相关生物学功能研究和药物发现。  内在无序蛋白及其无序区域在真核生物蛋白质组中非常普遍,约占人类蛋白质组的三分之一。这类蛋白并不形成稳定的三维结构,而是以快速相互转换的多种构象集合形式存在,其生物学功能往往依赖于这些构象系综的整体性质。例如,它们参与信号转导、基因表达调控、染色质组织、细胞周期控制以及生物分子凝聚体形成等重要过程。
由于无序蛋白的功能与其构象分布密切相关,仅依赖单一结构模型难以解释其行为,因此需要结合实验测量与计算模拟来生成和分析构象系综。为了解决当前方法分散、标准不统一的问题,研究人员提出建立统一的分析框架,以便在不同实验技术和计算模型之间实现一致的解释和验证。
方法概述:统一框架的总体思想 该统一框架围绕三个核心步骤展开:首先获取能够反映构象分布的实验数据,其次利用计算方法生成符合实验约束的构象系综,最后通过独立数据和统计方法对结果进行验证与比较。该框架强调实验与计算的循环迭代过程,即实验数据指导模型生成,而模型又用于解释实验结果,从而逐步逼近真实的构象分布。
在这一框架中,实验模块提供局部或整体结构信息,计算模块负责生成大量可能的构象集合,而验证模块则通过统计距离、结构合理性和功能相关性等指标判断模型是否可靠。通过这种模块化设计,不同研究团队可以根据具体问题选择合适的实验与计算策略,同时保持整体流程的一致性。 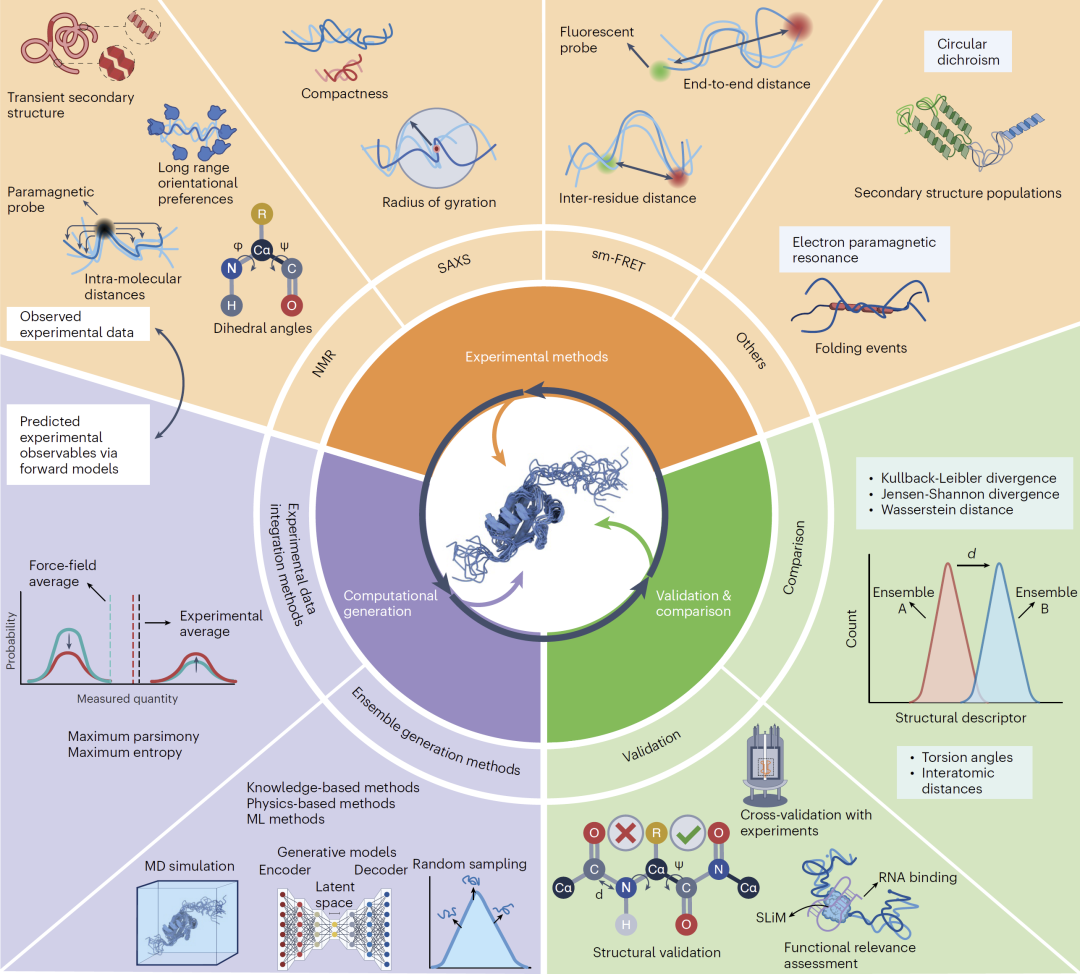 图1:用于确定无序蛋白构象系综的模块化框架。
实验方法:获取无序蛋白构象信息 研究无序蛋白构象系综需要多种互补的实验技术,其中核磁共振是最重要的方法之一。该技术可以在溶液中提供原子分辨率的信息,并能够反映蛋白在不同时间尺度上的动态行为。然而,核磁信号通常是所有构象的平均值,因此需要结合计算模型才能解释具体结构。
单分子荧光共振能量转移技术可以测量分子内部距离分布,从而揭示不同构象之间的变化。小角散射技术则提供蛋白整体尺寸和形状信息,对于描述无序蛋白的扩展或压缩状态非常重要。除此之外,氢氘交换、圆二色谱和电子顺磁共振等方法也可以提供关于局部结构、柔性以及瞬时接触的信息。
由于每种实验技术只能提供部分信息,因此需要将多种数据结合使用,才能得到可靠的构象系综描述。
计算方法:生成构象系综 构象系综的生成通常依赖三类计算策略。第一类是基于已知结构统计规律的方法,通过片段库或二面角分布快速生成大量可能构象;第二类是基于物理原理的分子动力学模拟,通过力场计算蛋白在不同条件下的运动轨迹;第三类是近年来兴起的机器学习方法,它们能够从已有数据中学习序列与构象之间的关系,并快速生成合理的结构集合。
为了使模拟结果与实验一致,通常需要将实验数据引入模型进行约束。常见策略包括最大熵方法,通过在保持构象多样性的同时满足实验条件来调整构象权重,以及最简原则方法,通过选择最少数量的构象来解释实验数据。这些方法能够在保持物理合理性的同时提高模型与实验的一致性。 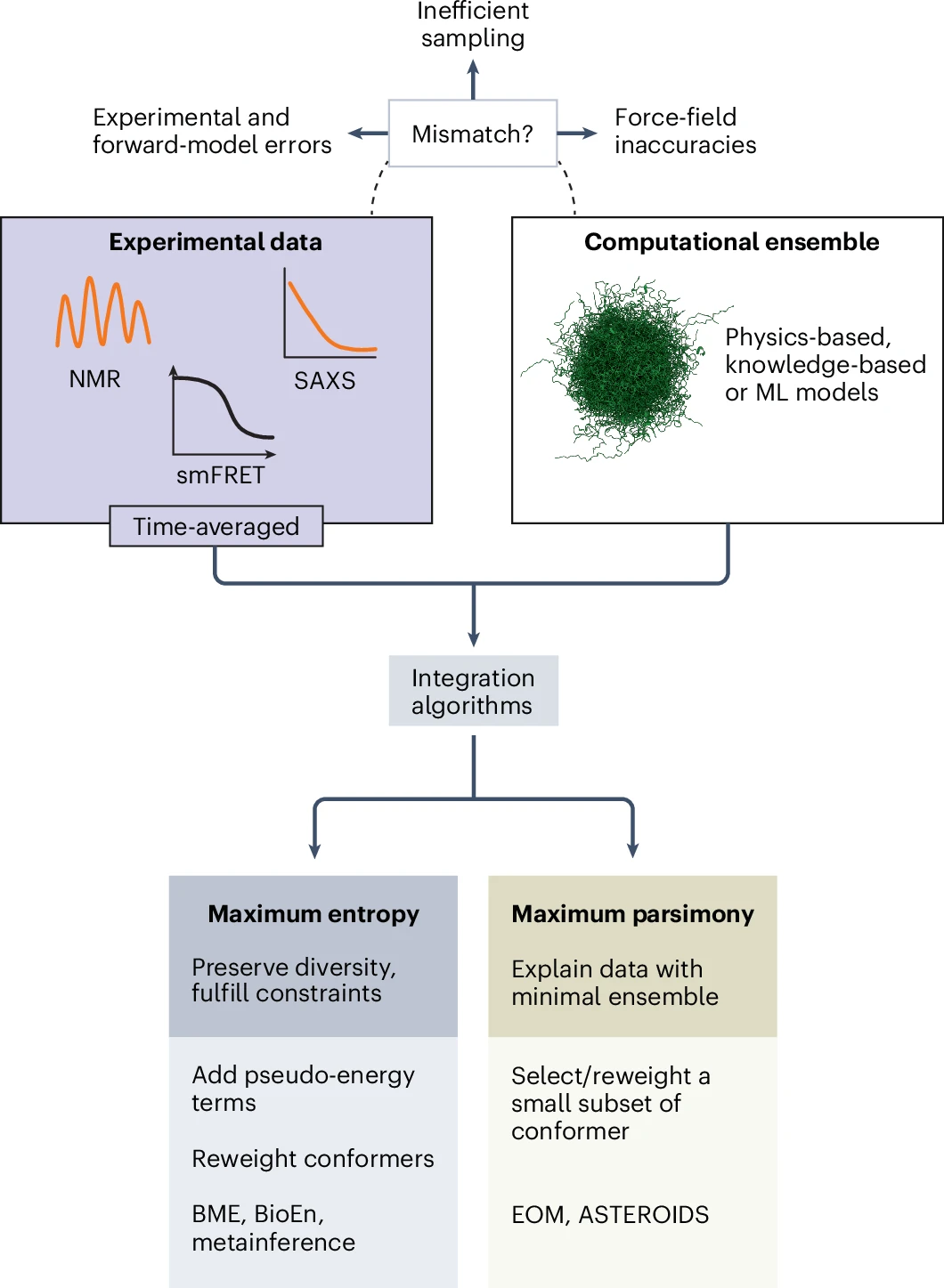 图2:无序蛋白构象系综整合建模策略概览。
构象系综的比较与验证 由于无序蛋白的构象空间非常庞大,通常会存在多种不同的模型能够解释同一组实验数据。因此,对构象系综进行比较和验证是不可或缺的步骤。
验证过程通常从基本的结构合理性检查开始,例如键长、键角和构象分布是否符合物理规律。随后需要检验模型是否能够再现实验测量得到的整体尺寸、距离分布和二级结构含量。进一步的验证包括利用独立实验数据进行交叉检验,以及评估构象是否能够解释蛋白的生物学功能。
此外,真实细胞环境中的构象可能与体外实验不同,因此未来的发展方向之一是利用细胞内实验数据来验证模型,从而得到更加真实的构象系综。
讨论 研究人员认为,无序蛋白构象系综的确定正处于关键阶段。虽然实验技术和计算方法发展迅速,但缺乏统一标准限制了不同研究结果之间的可比性。因此,建立统一框架并开展社区级基准测试,将有助于推动该领域的发展。
未来的重要方向包括提高力场精度、改进采样效率、发展更可靠的实验数据解释模型,以及引入不确定性评估机制。此外,还需要考虑无序蛋白的动力学行为和非平衡过程,因为许多功能依赖于瞬时构象变化而不是静态结构。
通过实验与计算的深度融合,并建立标准化流程,研究人员希望能够实现对无序蛋白构象空间的系统理解,从而推动基础生物学研究和药物设计的发展。 整理 | DrugOne团队
参考资料
Ghafouri, H., Kadeřávek, P., Melo, A.M. et al. Toward a unified framework for determining conformational ensembles of disordered proteins. Nat Methods (2026). https://doi.org/10.1038/s41592-026-03003-2
|
 关注公众号
关注公众号
 发表于 2026-3-19 23:37:25
发表于 2026-3-19 23:37:25
